
alport syndrome
Alport syndrome is a rare kidney disease that is genetically passed down in families from parents to children, though sometimes it can occur spontaneously and you might be the first one in your family to have it.
The disease causes one’s kidney function to decline over time and can also result in hearing loss and eye abnormalities. A small percentage of X-linked Alport syndrome patients experience leiomyomas, benign (not cancerous) smooth muscle tumors that can be found in the esophagus, lungs, uterus, and other female reproductive organs. The acronym ASDL is commonly used to refer to Alport syndrome with diffuse leiomyomatosis.
Prevalence is estimated to be less than 200,000 people in the U.S. making Alport syndrome a rare disease. No reliable prevalence studies are available.
The current goal of all treatment is to help slow progression of Alport syndrome so that patients keep their “native” kidneys as long as possible.
Blood-pressure lowering medicines are often prescribed for this reason, even if patients don’t have high blood pressure. ACE and ARB medications; Lisinopril, Ramipril, and Losartan – are proven to slow the spill of protein into the urine, slowing the scarring of the kidneys. Current research is in process to more fully understand the disease, identify more treatment options and a potential cure. Several human clinical trials are in progress as well.
There are three different genetic types of Alport syndrome.
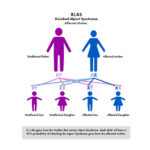
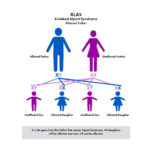
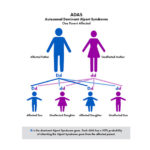
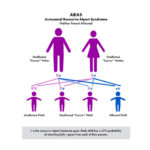
The most common is called X–linked (XLAS). Because males only have one X–chromosome (females have two), the disease can be harder on males and their kidney function can decline faster than females. The other genetic types are recessive Alport syndrome (ARAS) and autosomal dominant (ADAS).
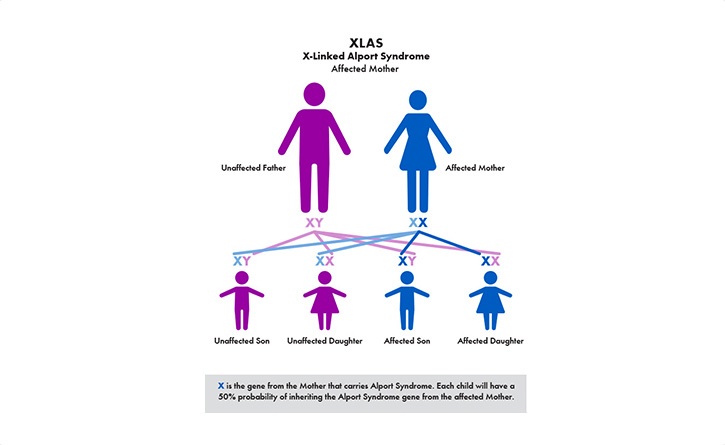
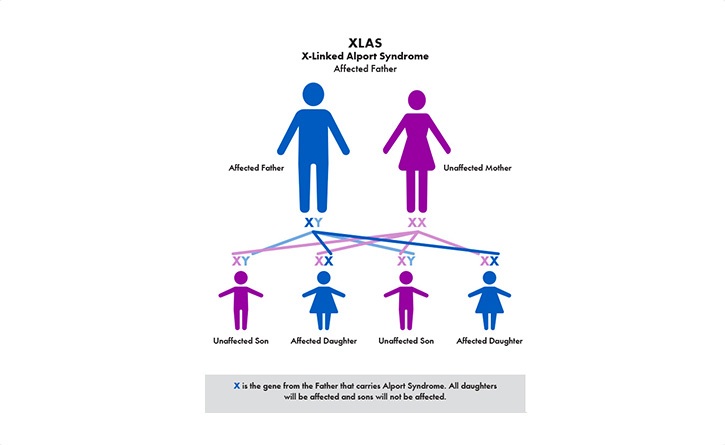
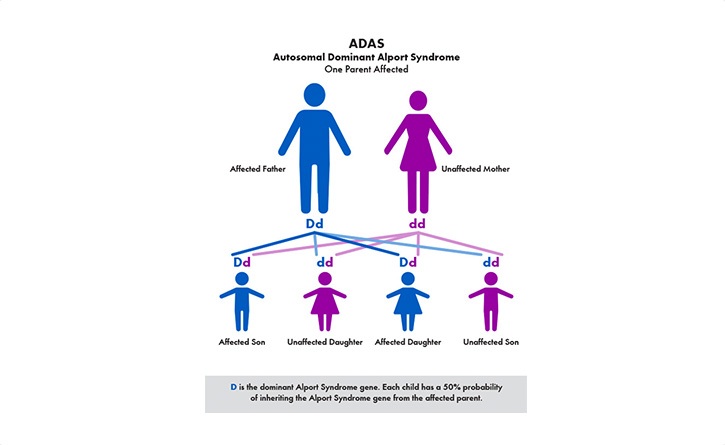
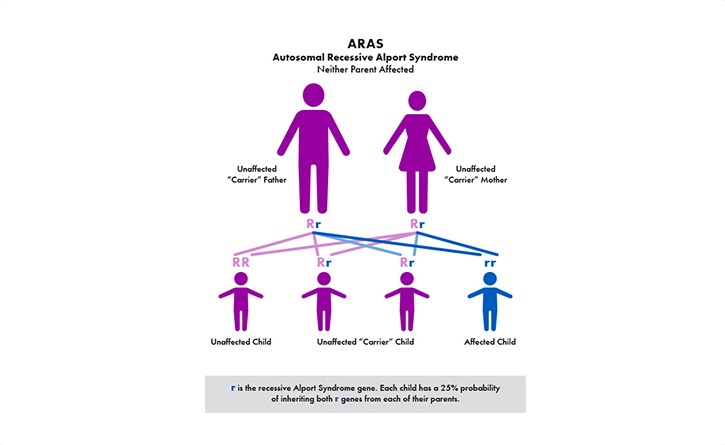
The primary symptom is blood in the urine (hematuria)
which is usually microscopic, meaning it can only be detected with a microscope or a urine dipstick. Sometimes children with Alport syndrome have brown, pink, or red urine (gross hematuria) for several days, associated with an infection. This gross hematuria stops when the child recovers from the infection, and can be alarming but is not harmful. As the disease progresses additional signs of kidney disease begin to appear, such as protein in the urine (proteinuria) and high blood pressure.
Alport syndrome is caused by genetic mutations that affect the type IV collagen family of proteins in the body.
Type IV collagen is a major part of important tissue structures called basement membranes that are present in all tissues including the kidney, inner ear, and eye. For this reason, Alport syndrome causes kidney disease, and in some people, it also causes hearing loss and vision problems. Some families do not experience hearing loss or vision problems at all.


NAMING THE SYNDROME
Alport syndrome is named after British doctor A. Cecil Alport, who in 1927 described three generations of a family in which multiple individuals exhibited progressive kidney disease and hearing loss.
Dr. Alport also observed that blood in the urine (hematuria) was the most common symptom and that males were affected more severely than females. After Dr. Alport’s observations, many more families were described and the disease was named Alport syndrome in 1961.